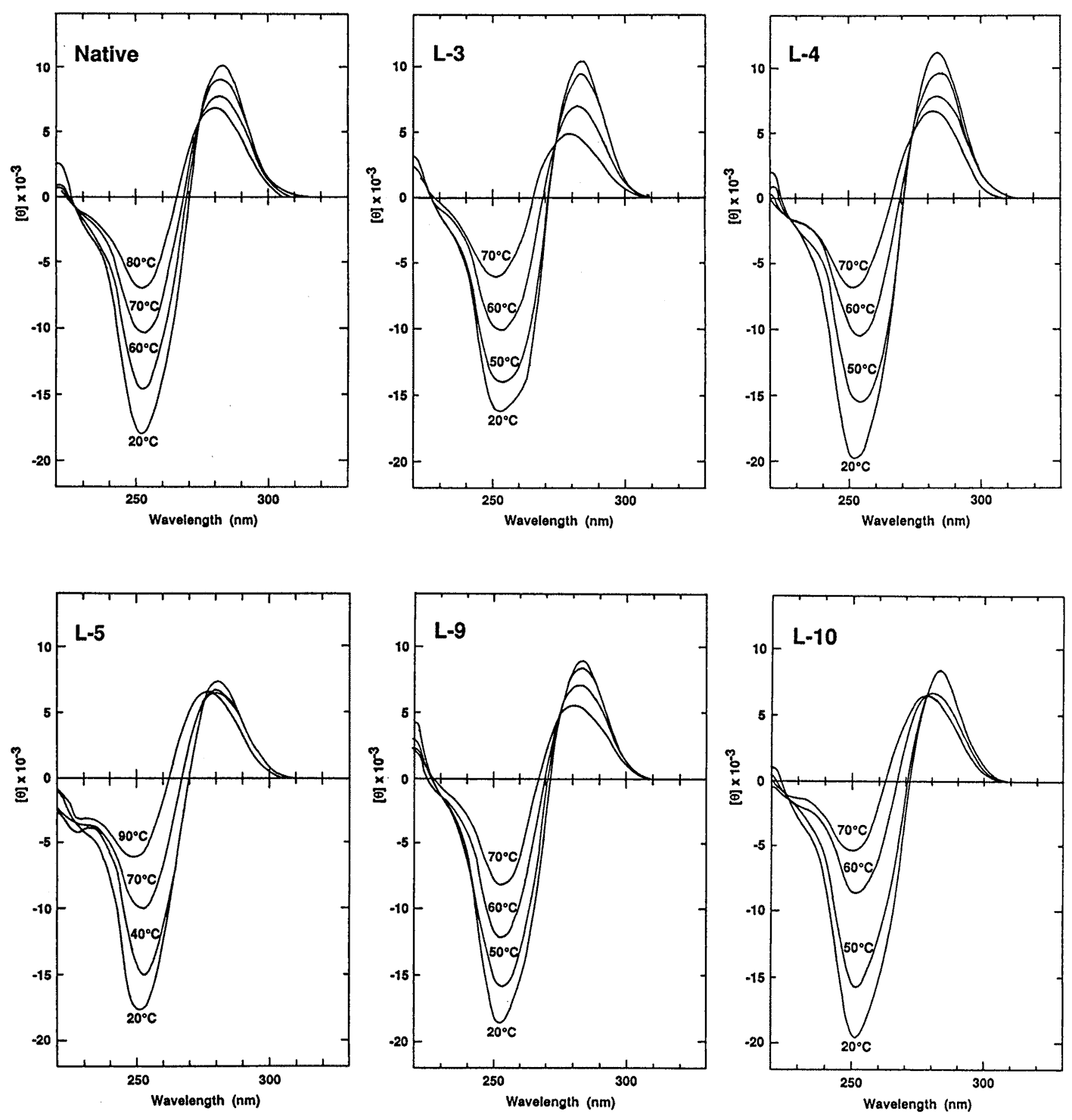
Figure 2. CD spectra of the native and heterochiral 12mers. Samples contained 8.5 mM strand in 1 M NaCl, 10 mM sodium phosphate, pH 7.5.
The macroscopic structural analyses of the 12mers provided the information for overall duplex structure of the 12mers. Our subsequent interest is how they form the duplex structure, especially at the substitution site with L-nucleotide. To obtain such information, we carried out nuclear magnetic resonance (NMR) experiments. The signal assignment was carried out by using the sequential assignment method which is widely applied to B-form DNA fragments [17]. Figure 3 shows the 1H NMR spectra of exchangeable imino resonance region of the native 12mer and L-4. Imino protons (guanine-1NH and thymine-3NH) serve as a proton donor for base pairing. Since imino protons, which are exchangeable with solvent protons, are unable to be detected when an oligonucleotide exists as a single strand. However, when the oligonucleotide forms a duplex (base pairing), the exchange rate of the imino protons dramatically reduces and the imino resonances become to be observed. In this case, the sequence of the 12mers has the twofold axis, and then only the protons in the region surrounded by a rectangle (Figure 4) are observable by 1H NMR. The native 12mer showed six imino resonances at 0°C (Figure 3, bottom), indicating all the base pairs to be formed. In the case of L-4, all imino resonances containing G4NH are also observed (Figure 3, upper), thus the unnatural G4 residue forms the base-pairing as well as other natural residues as expected by the UV melting experiments. Figure 5a and 5b shows the temperature dependence of the chemicalshifts for the imino resonances of L-4 and the native12mer, and the chemical shift differences of the non-exchangeable protons between L-4 and the native 12mer, respectively. The temperature dependence of the imino resonances is quite similar in both 12mers (Figure 5a). Imino protons in duplex DNA are positioned where are strongly affected by ring current effects of neighboring base pairs. Therefore, this result suggests that L-4 still retains the similar base pairing mode and base-base stacking geometry as parental one. In the case of the non-exchangeable protons, the large chemical shift differences between L-4 and the native 12mer are converged at the G4 residue (Figure 5b), and therefore the introduction of L-deoxyguanosine into the G4 site hardly affects the structure of the other regions. These results suggest that L-4 still retains the B-form type base pairing and base stacking, and the introduction of L-deoxyguanosine does not significantly affect the overall duplex structure.
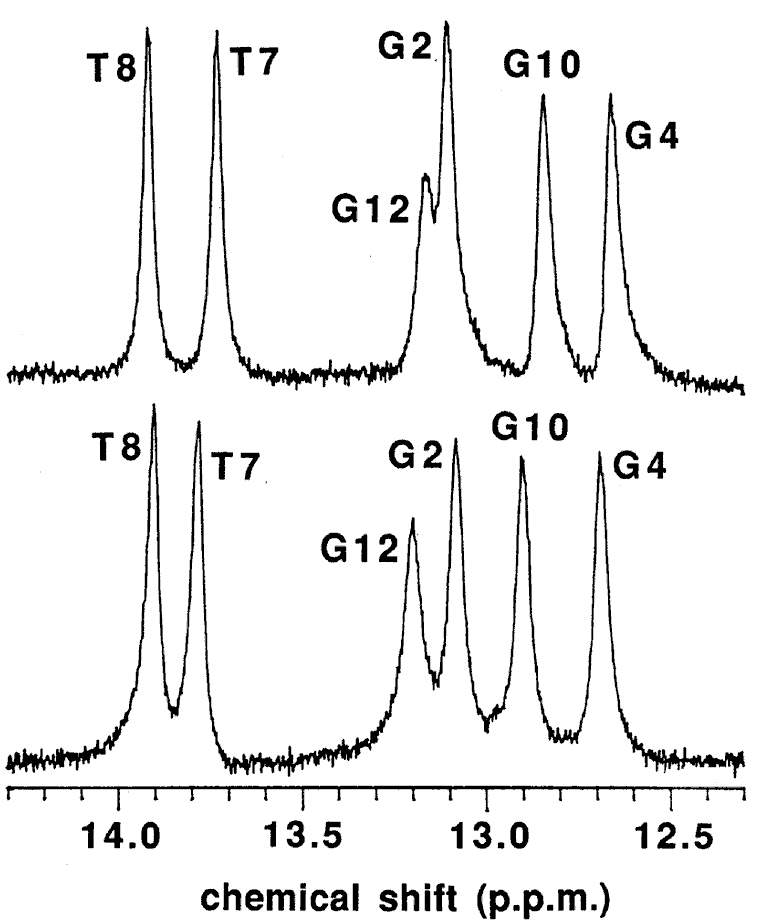
Figure 3. 1H NMR spectra of L-4 (upper) and the native 12mer (bottom) in the exchangeable imino resonance region. Samples contained 2.5 mM duplex in 0.1 M NaCl, 0.1 mM EDTA, 10 mM sodium phosphate (pH 7.5) in 20 % D2O at 1°C.
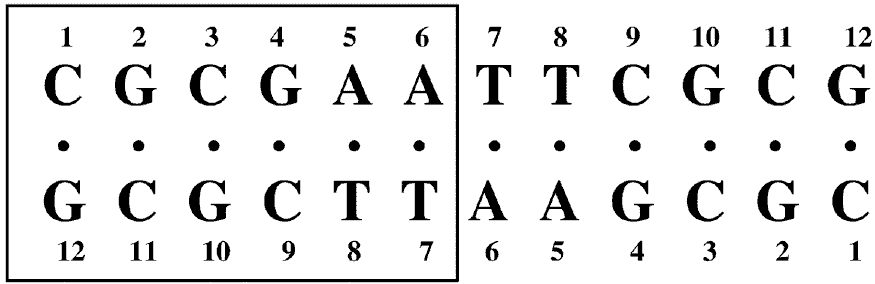
Figure 4. Nucleotide sequence and numbering system of the 12mer duplexes. The G4 residue of L-4 is unnatural L-deoxyguanosine. Since this sequence has a dyad axis, only protons in the residues surrounded by a rectangle can be observed.
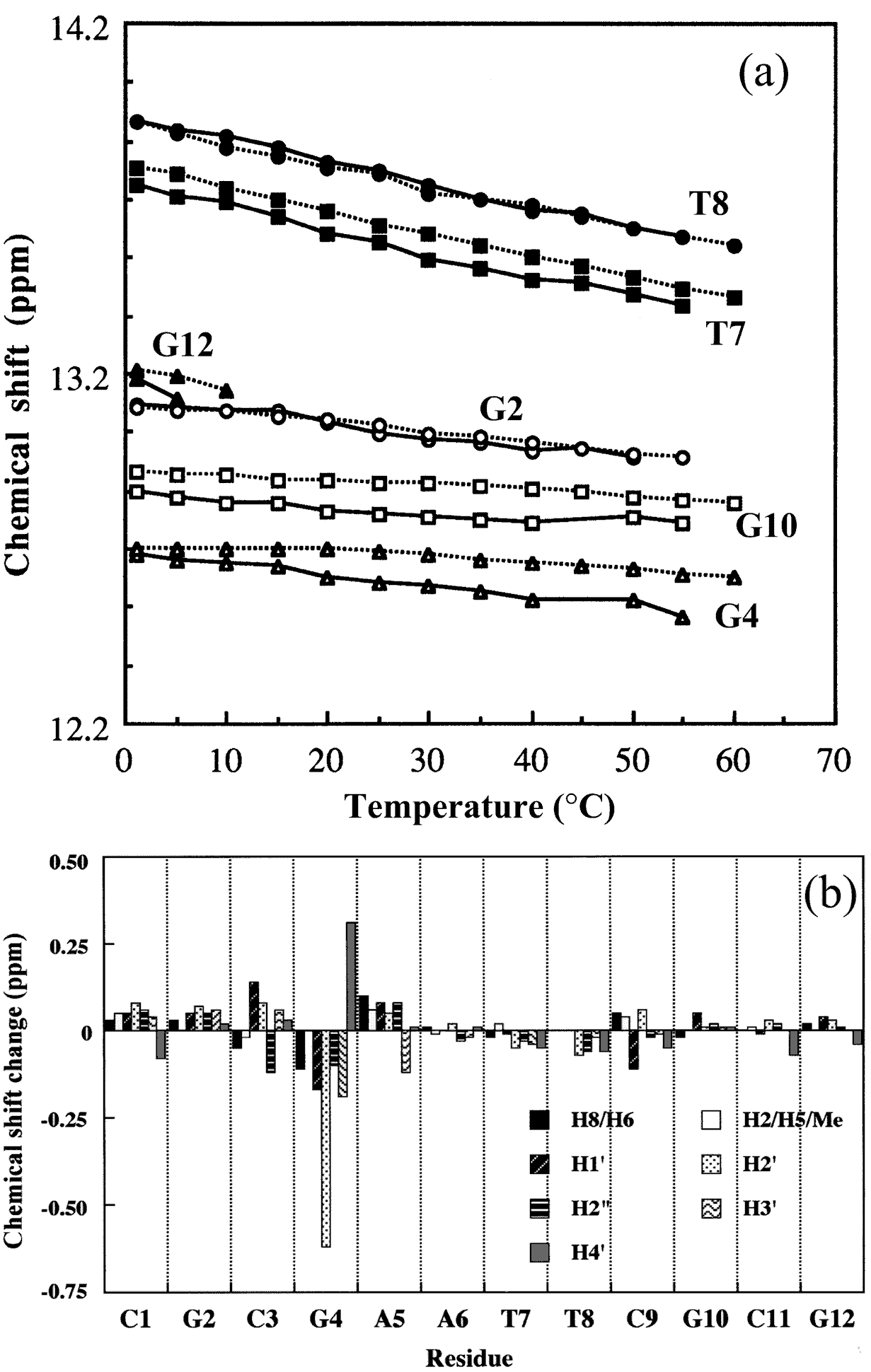
Figure 5. (a) Temperature dependence of chemical shifts of imino resonances of L-4 (solid line) and the native 12mer (dotted line). (b) Chemical shift differences between L-4 and the native 12mer at 25°C. The differences are defined by Δδd = δ (L-4)−δ (native 12mer).
<
Figure 6a shows the sequential NOE connectivities between the base H8 (purines) or H6 (pyrimidines) protons and sugar H1' protons of L-4. As shown in Figure 6a, the NOE cross-peaks could be completely connected. This means that the duplex structure of the 12mer does not change dramatically by the substitution of the G4 residue with L-deoxyguanosine. Figure 6b shows the sequential NOE connectivities between the base H8 or H6 protons and sugar H3' protons of L-4. In this case, the NOEs derived from G4H8-G4H3' and A5H8-G4H3' are missing, instead, unusual NOEs derived from G4H8-G4H4' and A5H8-G4H4' were observed. It was found that all the residues in L-4 including the G4 residue adopt the S-type sugar puckering by the analysis of the DQF-COSY spectrum (data not shown). When the deoxyguanosie residue with the S-type sugar conformation adopts a usual anti glycosyl conformation, strong NOEs between base H8 and the sugar H2', H3' are observed. In the case of the G4 residue of L-4, the NOE between H6 and H3' could not be observed as described above, but the weak NOE between H6 and H4' was observed. This means that H6 is close to rather H4' than H3' [3]. This result indicates that the unnatural G4 residue of L-4 adopts an unusual low anti glycosyl conformation (χ = 180° ± 30°) (χ is defined by a torsion angle of C4-N9-C1'-O4' for prines and C2-N1-C1'-O4' for pyrimidines), although each residue of regular B-form DNA fragments adopts an anti glycosyl conformation (χ = 135° ± 30°) (Figure 7).
Based on the above experimental results, the model structure of L-4 was constructed. As shown in Figure 8, the G4 residue forms the Watson-Crick base-pairing with the complementary C9 residue. Each O4' atom of the sugar moiety in the regular B-form DNA is oriented toward the 5'-end direction like as the C3 and A5 residues in L-4, however, that of the G4 residue is oriented toward the opposite direction. This characteristic conformation would make the L-deoxyguanosine residue enable to form the usual Watson-Crick base pairing.
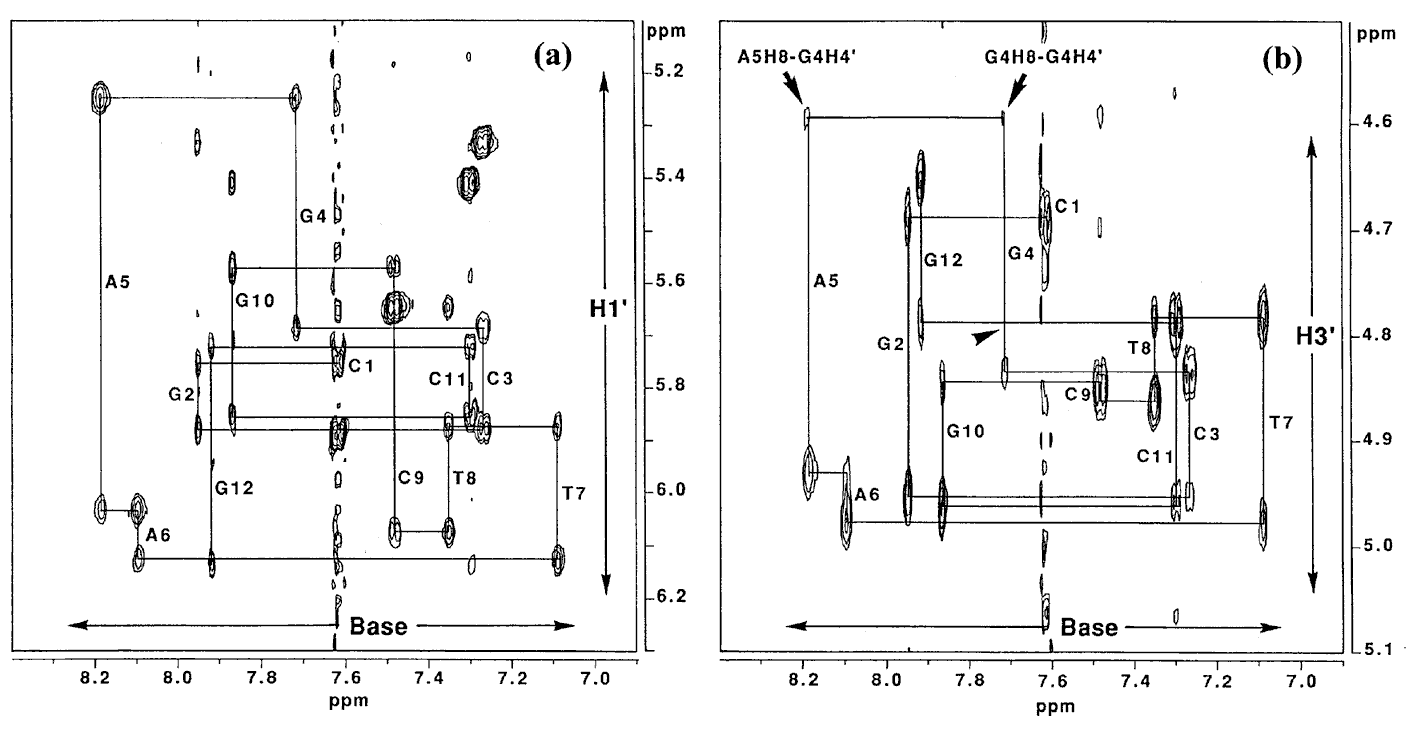
Figre6. NOESY spectrum (150 ms mixing time) of L-4 in D2O at 25°C. Duplex concentration is 2.5 mM in 0.1 M NaCl, 0.1 mM EDTA, 10 mM sodium phosphate, pD 7.5. (a) Expanded base proton and sugar H1' region of the spectrum. (b) Expanded base proton and sugar H3' region of the spectrum. Arrowhead represents the chemical shift of the missing G4H8-G4H3'.
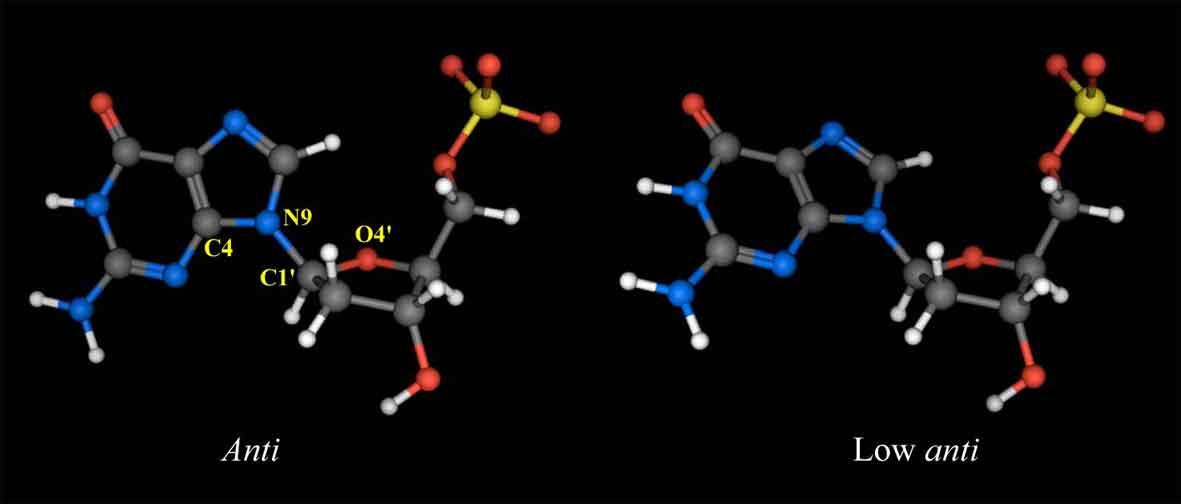
Figure 7. Structures of L-deoxyguanosine 5'-monophosphate with the anti (χ = 135°±30°, left) and low anti (χ = 180°±30°, right) glycosyl conformations. χ is defined by a torsion angle of C4-N9-C1'-O4'.
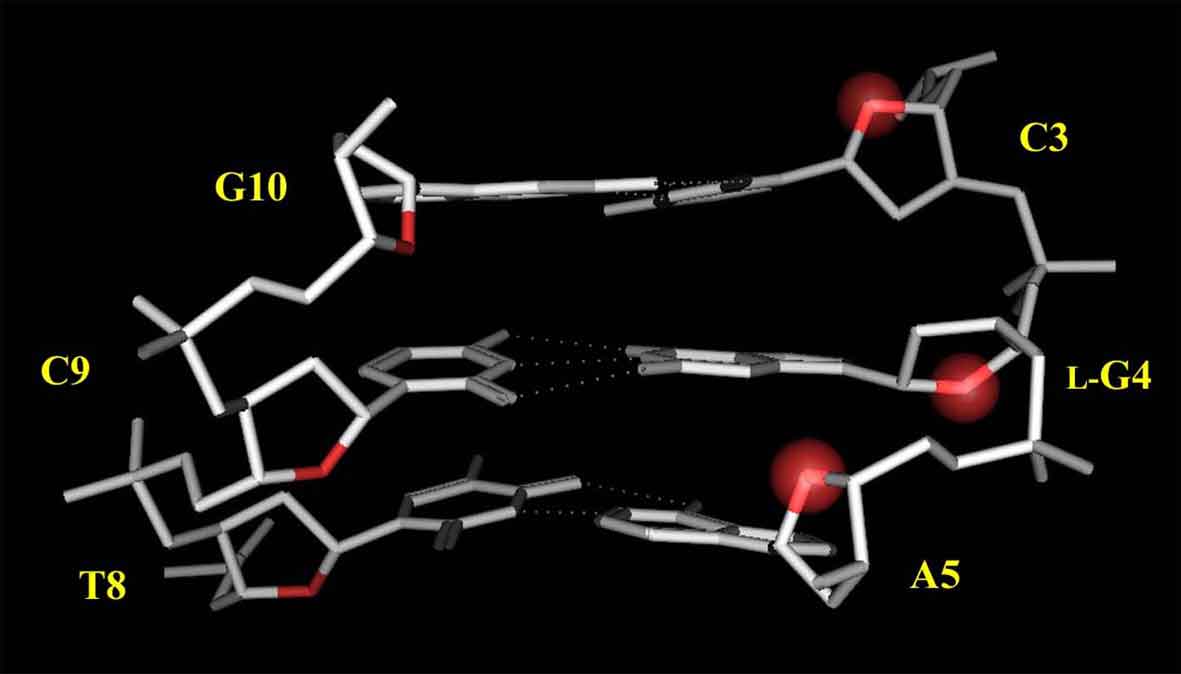
Figure 8. Model structure of L-4. Protons and the residues other than C3, L-G4, A5 are omitted for clarity. The O4' atoms of the C3, L-G4, A5 residues are highlighted with a red sphere.
To further evaluate the duplex structure of the 12mers, digestion experiments of the 12mers with restriction endonuclease EcoRI was carried out. This enzyme strictly recognizes the sequence of 5'-GAATTC-3' in double-stranded B-form DNA and cleaves a phosphodiester bond at the GpA step in the recognition sequence. The specificity of the DNA recognition by this enzyme is achieved by both direct and indirect read-out mechanisms [18,19], which involve protein-base contacts through hydrogen-bonding and van der Waals contacts (direct read-out), and recognition of the overall conformation of DNA (indirect read-out). Therefore, the susceptibility of DNA fragments for the restriction endonuclease digestion affords structural information of the substrate DNAs. The time-course of the cleavage reaction of the 12mers with EcoRI is shown in Figure 9. The reaction was performed at 20°C, since these 12mers form the stable duplex at this temperature from the CD and the UV melting experiments. Under the conditions that native 12-mer is completely hydrolyzed (48 hr), L-4 and L-5 show complete resistance to digestion with this enzyme. In contrast, L-3, L-10 and even L-9 that contains the L-nucleotide in the recognition sequence were hydrolyzed, although the cleavage rate of L-3 and L-9 is significantly reduced. Surprisingly, the cleavage rate of L-10 is about 20-fold enhanced compared with that of the native 12mer. From the CD experiments, the overall duplex structure of L-10 has significantly different characteristics from that of the other 12mers. The enhanced hydrolysis rate of L-10 would be due to the indirect effect such as conformational change, since the direct interaction between EcoRI and the 3'-flanking residue of the recognition sequence, corresponding to the L-deoxyguanosine residue in L-10, was not observed in the co-crystal of the EcoRI-DNA fragment complex [19]. Thus, L-10 might have the kinked structure to easily accommodate the binding with EcoRI as seen in the EcoRI-DNA fragment complex. The results obtained for L-4 and L-5 may suggest that the introduction of L-nucleotide at the catalytic site is critical for the reaction (catalysis). Thus, heterochiral 12mers retains the B-form type duplex structure that can be recognized by EcoRI.
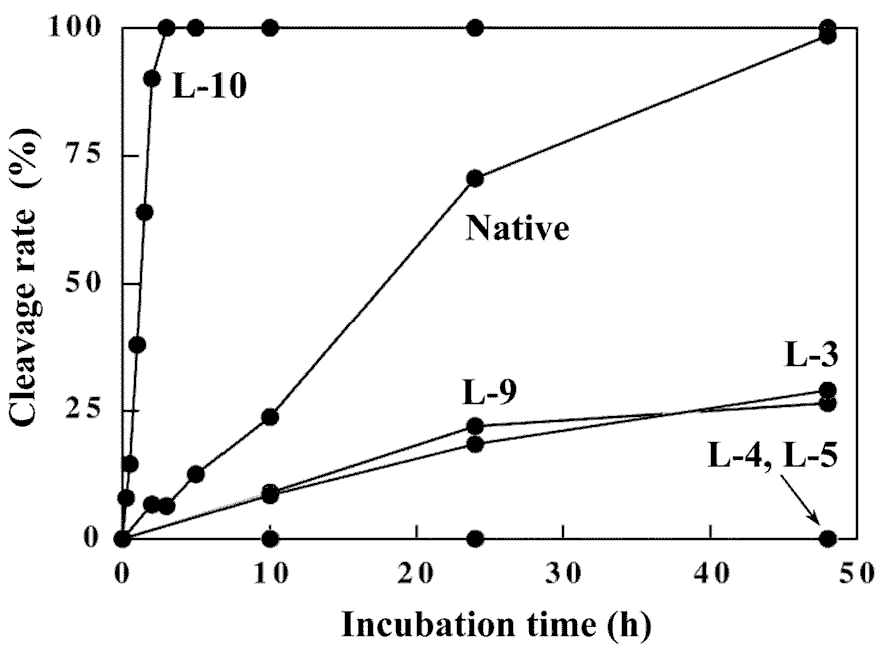
Figure 9. Time-course of cleavage reaction of the 12mers with restriction endonuclease EcoRI.
The establishment of the homochirality of nucleic acids and proteins is thought to have been essential for the origins of life. The chemical processes that biomolecules acquire the homochirality can be considered to involve three events: 1) symmetry breaking, 2) chiral amplification, and 3) chiral transfer. Symmetry breaking is explained by, for example, a predicted small energy difference between enantiomeric molecules as a result of a parity violation in the weak nuclear force [20], or a merely statistical chance [21]. Chiral amplification can make molecules possible to achieve large enantiomeric excesses from small chiral imbalance [22]. The amplified chirality of a molecule transfers to other molecules (chiral transer). Since the rate of racemization of amino acids is relatively too fast, it would not be reasonable to consider the chirality of amino acids and peptides as a source of the chirality of the biosphere. We have thus focused on the chirality of nucleic acids. Our results described here indicate that the introduction of an L-nucleotide into DNA does not induce significant helical distortions. In other words, the DNA double-helical structure permits the incorporation of a chiral antipode, although some decreases of the duplex stability are observed. This allows us to speculate that heterochiral DNA can serve as a template for the non-enzymatic oligomerization. However, the decrease of the duplex stability would serve unfavorably for the complex formation of a template with monomers and growing strands, and for the chemical stability of DNA [23]. Actually, Orgel and coworkers reported that the non-enzymatic oligomerization of activated guanosine 5'-monophosphate (2-MeImpG) on a DNA template containing one or two successive L-deoxycytidine residues proceeds to the end of the template through the L-deoxycytidine residues [24]. However, the efficiency is rather low relative to the oligomerization on an all-D-template, furthermore a template containing three successive L- deoxycytidine residues or alternating D-,L-template does not facilitate the oligomerization. The structural characteristics of heterochiral DNA shown in this study well explain the template activity of the DNA oligomers containing L-nucleotides reported by Orgel et al [24]. Overall, the introduction of L-type monomers into DNA should be unfavorable for the non-enzymatic oligomerization and replication of DNA. Such characteristic of heterochiral DNA might have in part contributed to the natural selection of homochiral DNA.
Conclusions
We have investigated the duplex structure of heterochiral DNA 12mers that have an unnatural L-nucleotide residue, and found that the breaking homochirality of the 12mer does not cause a drastic structural alteration. However, the breaking homochirality of the 12mer leads to the some destabilization of the duplex. Such destabilization might have been disadvantageous for the natural selection of heterochiral DNAs in the processes of the chemical evolution of DNA via oligomerization of mononucleotides that would have been abiotically synthesized as a racemate on the primitive earth.Acknowledgments:
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture, Japan.References
[1] Joyce, G. F., Schwartz, A. W., Miller, S. L. and Orgel, L. E. The case for an ancestral genetic system involving simple analogs of the nucleotides, Proc. Natl. Acad. Sci. U.S.A., 84, 4398-4402 (1987).[2] Urata, H., Shinohara, K., Ogura, E., Ueda, Y. and Akagi, M. Mirror Image DNA, J. Am. Chem. Soc. 113, 8174-8175 (1991).
[3] Urata, H., Ueda, Y., Suhara, H., Nishioka, E. and Akagi, M. NMR study of a heterochiral DNA: stable Watson-Crick-type base-pairing between the enantiomeric residues, J. Am. Chem. Soc. 115, 9852-9853 (1993).
[4] Blommers, M. J. J., Tondelli, L. and Garbesi, A. Effects of the introduction of L-nucleotides into DNA. Solution structure of the heterochiral duplex d(G-C-G-(L)T-G-C-G)・d(C-G-C-A-C-G-C) studied by NMR spectroscopy, Biochemistry 33, 7886-7896 (1994). [5] Fujimori, S., Shudo, K. and Hashimoto, Y. Enantio-DNA recognizes complementary RNA but not complementary DNA, J. Am. Chem. Soc. 112, 7436-7438 (1990).
[6] Asseline, U., Hau, J.-F., Czernecki, S., Diguarher, T. L., Perlat, M.-C., Valery, J.-M. and Thuong, N. T. Synthesis and physicochemical properties of oligonucleotides built with either a-L or b-L nucleotides units and covalently linked to an acridine derivative, Nucleic Acids Res. 19, 4067-4074 (1991).
[7] Anderson, D. J., Reischer, R. J., Taylor, A. J. and Wechter, W. J. Preparation and characterization of oligonucleotides of D- and L-2'-deoxyuridine, Nucleosides Nucleotides 3, 499-512 (1984).
[8] Morvan, F., Genu, C., Rayner, B., Gosselin, G. and Imbach, J.-L. Synthesis, nuclease resistance and base pairing properties of α- and β-L-octathymidylates, Biochem. Biophys. Chem. Res. 172, 537-543 (1990).
[9] Garbesi, A., Capobianco, M. L., Colonna, F. P., Tondelli, L., Arcamone, F., Manzini, G., Hilbers, C. W., Aelen, J. M. E. and Blommers, M. J. J. L-DNAs as potential antimessenger oligonucleotides: A reassessment, Nucleic Acids Res. 21, 4159-4165 (1993).
[10] Damha, M. J., Giannaris, P. A. and Marfey, P. Antisense L/D-oligodeoxynucleotide chimeras: Nuclease stability, base-pairing properties, and activity at directing ribonuclease H, Biochemistry 33, 7877-7885 (1994).
[11] Urata, H. and Akagi, M. Sequence dependence of thermodynamic stability of heterochiral DNA, Tetrahedron Lett. 37, 5551-5554 (1996).
[12] Urata, H., Ogura, E., Shinohara, K., Ueda, Y. and Akagi, M. Synthesis and properties of mirror-image DNA, Nucleic Acids Res. 20, 3325-3332 (1992).
[13] States, D. J., Haberkorn, R. A. and Ruben, D. J. A two-dimensional nuclear Overhauser experiment with pure absorption phase in four quadrants, J. Magn. Reson. 48, 286-292 (1982).
[14] Fasman, G. D. Ed., Handbook of biochemistry and Molecular Biology, 3rd ed., Vol. 1, Nucleic Acids; pp. 589, CRC Press, Boca Raton, FL, 1975.
[15] Marky, L. A., Blumenfeld, K. S., Kozlowski, S. and Breslauer, K. J. Salt-dependent conformational transitions in the self-complementary deoxydodecanucleotide d(CGCGAATTCGCG): evidence for hairpin formation, Biopolymers 22, 1247-1257 (1983).
[16] Leonard, G. A., Booth, E. D. and Brown, T. Structural and thermodynamic studies on the adenineラguanine mismatch in B-DNA, Nucleic Acids Res. 18, 5617-5623 (1990).
[17] Hare, D. R., Wemmer, D. E., Chou, S. H., Drobny, G. and Reid, B. R. Assignment of the nonexchangeable proton resonances of d(C-G-C-G-A-A-T-T-C-G-C-G) using two-dimensional nuclear magnetic resonance methods, J. Mol. Biol. 171, 319-336 (1983).
[18] Lesser, D. R., Kurpiewski, M. R. and Jen-Jacobson, L. The energetic basis of specificity in the Eco RI endonuclease-DNA interaction, Science 250, 776-786 (1990).
[19] McClarin, J. A., Frederick, C. A., Wang, B.-C., Greene, P., Boyer, H. W., Grable, J. and Rosenberg, J. M. Structure of the DNA-Eco RI endonuclease recognition complex at 3 Å resolution, Science 234, 1526-1541 (1986).
[20] Wu, C. S., Ambler, E., Hayward, R. W., Hoppes, D. D. and Hudson, R. P. Experimental test of parity conservation in β-decay, Physical Review 105, 1413-1415 (1957). [21] Thiemann, W. The origin of optical activity, Naturwissenschaften 61, 476-483 (1974).
[22] Soai, K., Shibata, T., Morioka, H. and Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule, Nature 378, 767-768 (1995).
[23] Lindahl, T. Instability and decay of the primary structure of DNA, Nature 362, 709-715 (1993).
[24] Kozlov, I. A., Pitsch, S. and Orgel, L. E. Oligomerization of activated D- and L-guanosine mononucleotides on templates containing D- and L-deoxycytidylate residues, Proc. Natl. Acad. Sci. USA 95, 13448-13452 (1988).